10-Oct-2023
In recent years, the field of structural biology has undergone a revolution thanks to the exceptional predicting capabilities of AI models, such as AlphaFold. With these tools, predicting the 3D structure of proteins has become quicker, and more accurate than ever before. Despite the accuracy of structure prediction, understanding how these proteins can interact with ligands or protein partners is not clear from these models. Although extra tools have been developed, such as AlphaFill, which does indeed take this next step in modelling co-factors, and protein metal binding sites for example, however experimental methods are still heavily relied upon for predicting the interaction of drug like compound chemical entities.
However, a team from Diamond Light Source and IBM wanted to determine whether generative models could be used to screen potential inhibitors of proteins targets, specifically for SARS-CoV-2 in this study. Using deep generative framework from IBM, the team whittled down hundreds of thousands of potential inhibitors of the receptor binding domain (RBD) of the Spike protein (S), and the primary protease Mpro.
The study (Chenthamarakshan et al, 2023) aimed to demonstrate that these generative models could discover previously unidentified protein inhibitors. The model was given a foundational knowledge of molecules, just by feeding it strings of text on protein structures and their binding capabilities. Crucially, the model was only provided with the basic information (amino acid sequences) of the structures of S and Mpro, in order to more robustly illustrate that the generated inhibitors were free from biases.
More than 875,000 candidates were identified in three days. These were then assessed with a range of criteria: affinity of binding to the target protein, toxicity and off-target binding which could cause adverse effects, candidates where the binding mechanism was unknown but still could inhibit the protein, and candidates that were feasible to synthesise. Roughly 100 candidates for each protein were identified.
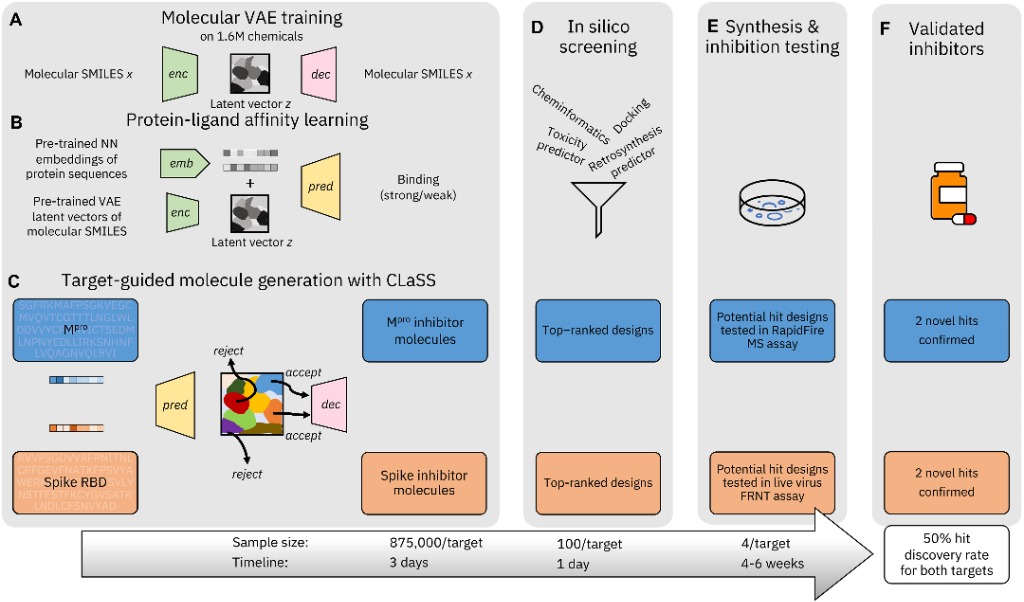
Figure 1. The process of generative modelling to sift through thousands of target inhibitors to 100, and finally to four per target through in silico screening. Of the four, two previously unknown successful inhibitors were identified.
In silico screening, with input from IBM and the chemical manufacturers, reduced the number then down to four for each protein. The overall process is shown above in Figure 1. The time taken to synthesise four potential inhibitors of the proteins was roughly one week – down from the months required otherwise.
To assess the compounds for inhibition, assays were carried out at the University of Oxford and macromolecular crystallography was conducted at Diamond Light Source to elucidate the structures. It found that for each target, two of the four candidates demonstrated substantial inhibition, leading to a success rate of 50% for obtaining novel compounds that inhibited both targets.
The compounds referred to as GEN725 and GEN727 in the published paper were shown to bind to the spike RBD and inhibit SARS-CoV-2 across all the main variants of concern. Even more excitingly, both candidates actually bind to tyrosines and hydrophobic residues in the lipid-binding pocket of the RBD (unlike many neutralising antibodies, which tend to target the ACE2 binding domain), which is conserved across several coronaviruses and variants of concern of SARS-CoV-2.
Of the Mpro candidates, after triage to four compounds again resulted in a 50% success rate that yielded two inhibitors, GXA70 and GXA112. Unlike for the spike RBD inhibitors, the Mpro pair bind to very different regions of the target protein; GXA112 forms hydrogen binds primarily with the P1 site of Mpro, whereas GXA70 mostly interacts with P2 residues. This, however, was identified with docking simulations, as experimental determination of the structure of Mpro bound to its inhibitors was not possible in this study.
What the study outlined was that the speed of accurately identifying potential compounds to accelerate the drug discovery process using AI. The distinct lack of information required by the generative model, and the success rate of identifying starting compounds for lead compound discovery (50% success for both), makes this a promising method for use in drug discovery workflows.
Furthermore, this provides a solid foundation for the ongoing work in the Fragment-Screen project, which brings together a variety of structural biology methods, as well as AI, to identify potential drug candidates in fragment-based drug discovery. Diamond Light Source and IBM are both key members of the Fragment-Screen consortium, and their collaboration in this study is central to the potential outcomes of the project.
Instruct-ERIC is also involved in the AI4Life project, which aims to empower life science researchers to harness the full potential of Artificial Intelligence (AI) and Machine Learning (ML) methods for bioimage analysis. Find out more about AI4Life here.
